紫外—可见吸收光谱分析方法
的有关信息介绍如下:4.3.1.1 定性分析
无机元素的定性分析应用紫外—可见分光光度法比较少,主要采用原子发射光谱法或化学分析法。在有机化合物的定性分析鉴定及结构分析方面,由于紫外-可见吸收光谱较为简单,光谱信息少,特征性不强,并且不少简单官能团在近紫外光区及可见光区没有吸收或吸收很弱,在应用时也有较大的局限性。但是,这种方法可适用于不饱和有机化合物,尤其是共轭体系的鉴定,以此推断未知物的骨架结构。此外,还可配合红外光谱法、核磁共振波谱法和质谱法等常用的结构分析法进行定性鉴定和结构分析,不失为一种有利的辅助方法。
吸收光谱的形状、吸收峰的数目和位置及相应的摩尔吸光系数,是定性分析的光谱依据,而最大吸收波长λmax及相应的εmax是定性分析的最主要参数。比较法有标准物质比较法和标准谱图比较法两种。利用标准物质比较,在相同的测量条件下,测定和比较未知物与已知标准物的吸收光谱曲线,如果两者的光谱完全一致,则可以初步认为它们是同一类化合物;利用标准谱图或光谱数据比较,对于没有标准物质或标准物质难于得到的物质,此方法适用。
4.3.1.2 结构分析
紫外—可见分光光度法可以进行化合物某些基团的判别,共轭体系及构型、构象的判断。
(1)某些特征基团的判别
有机物的不少基团(生色团),如羰基、苯环、硝基、共轭体系等,都有其特征的紫外或可见光吸收带,紫外-可见分光光度法在判别这些基团时,有时是十分有用的。如在270~300nm处有弱的吸收带,且随溶剂极性增大而发生蓝移,就是羰基产生吸收带的有力证据;在184nm附近有强吸收带、204nm附近有中强吸收带、260nm附近有弱吸收带且有精细结构,则是苯环的特征吸收,等等。
(2)共轭体系的判断
共轭体系会产生很强的K吸收带,通过绘制吸收光谱,可以判断化合物是否存在共轭体系或共轭的程度。如果一化合物在210nm以上无强吸收带,可以认定该化合物不存在共轭体系;若215~250nm区域有强吸收带,则该化合物可能有两至三个双键的共轭体系,如1,3-丁二烯,λmax为217nm,εmax为21000;若260~350nm区域有很强的吸收带,则可能有三至五个双键的共轭体系,如癸五烯有五个共轭双键,λmax为335nm,εmax为118000。
(3)异构体的判断
包括顺反异构及互变异构两种情况的判断。
顺反异构体的判断:生色团和助色团处于同一平面时,会产生最大的共轭效应。由于反式异构体的空间位阻效应小,分子的平面性较好,共轭效应强,因此λmax及εmax都大于顺式异构体。
互变异构体的判断:某些有机化合物在溶液中可能有两种以上的互变异构体处于动态平衡中,这种异构体的互变过程常伴随有双键的移动及共轭体系的变化,因此会产生吸收光谱的变化。最常见的是某些含氧化合物的酮式与烯醇式异构体之间的互变。例如,乙酰乙酸乙酯就是酮式和烯醇式两种互变异构体,它们的吸收特性不同,酮式异构体在近紫外光区时λmax为272nm(εmax为16000);烯醇式异构体的λmax则为243nm(εmax为16000)。两种异构体的互变平衡与溶剂有密切关系,在像水这样的极性溶剂中,由于羰基可能与H2O形成氢键以降低能量达到稳定状态,所以酮式异构体占优势;而在像乙烷这样的非极性溶剂中,则形成分子内的氢键且形成共轭体系,以使能量降低达到稳定状态,所以烯醇式异构体比率上升。
此外,紫外—可见分光光度法还可以判断某些化合物的构象(如取代基是平伏键还是直立键)及旋光异构体等。
4.3.1.3 定量分析
紫外—可见分光光度法定量分析的常见方法有以下几种。
(1)单组分的定量分析
如果在一个试样中只要测定一种组分,且在选定的测量波长下,试样中其他组分对该组分不干扰,那么进行单组分的定量分析较为简单。一般有标准对照法和标准曲线法两种。
标准对照法:在相同条件下,平行测定试样溶液和某一浓度cS(应与试液浓度接近)的标准溶液的吸光度Ax和AS,则由cS可计算出试样溶液中被测物质的浓度cx。
AS=KcS,Ax=Kcx,cx=cSAx/AS
由于标准对照法仅使用单个标准,引起误差的偶然因素较多,故结果往往较不可靠。
标准曲线法:是实际分析工作中最常用的一种方法。配制一系列不同浓度的标准溶液,以不含被测组分的空白溶液作为参比,测定标准系列溶液的吸光度,绘制吸光度-浓度曲线,称为校准曲线(包括标准曲线或工作曲线)。在相同条件下测定试样溶液的吸光度,从校准曲线上找出与之对应的未知组分的浓度。
此外,有时还可以采用标准加入法(做法与原子吸收光谱法相同)。
(2)多组分的定量分析
根据吸光度具有加和性的特点,在同一试样中可以同时测定两种或两种以上的组分。假设要测定试样中的两种组分为A、B,如果分别绘制A、B两纯物质的吸收光谱,可能有三种情况,如图4.12所示。图4.12 a表明两组分互不干扰,可以用测定单组分的方法分别在λ1、λ2测定A、B两种组分;图4.12 b表明A组分对B组分的测定有干扰,而B组分对A组分的测定无干扰,则可以在λ1处单独测量A组分,求得A组分的浓度cA,然后在λ2处测量溶液的吸光度及A、B纯物质的和,根据吸光度的加和性则可以求出cB;图4.12c表明两组分彼此互相干扰,此时在λ1、λ2处分别测定溶液的吸光度
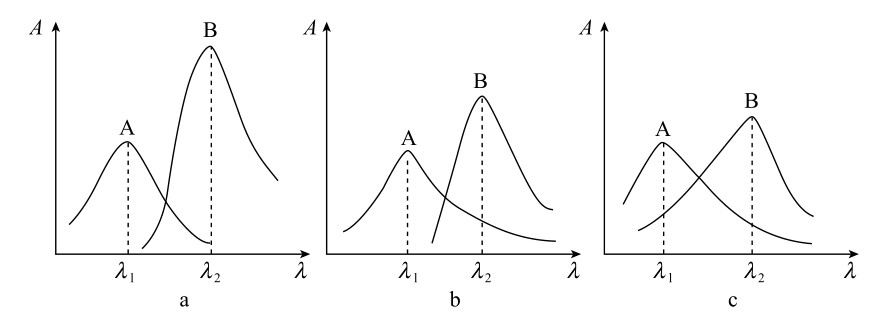
图4.12 混合物的紫外吸收光谱
图4.12 混合物的紫外吸收光谱
a—不重叠;b—部分重叠;c—相互重叠
显然,如果有n个组分的光谱互相干扰,就必须在n个波长处分别测定吸光度的加和值,然后解n元一次方程以求出各组分的浓度。应该指出,这是一个烦琐的数学处理过程,且n越多,结果的准确性越差,如果使用计算机进行测定结果的处理将使运算大大简便。
(3)双波长分光光度法
当试样中两组分的吸收光谱重叠较为严重时,用解联立方程的方法测定两组分的含量可能误差较大,这时可以用双波长分光光度法测定。它可以进行在有其他组分干扰的情况下测定某一组分的含量,也可以同时测定两组分的含量。双波长分光光度法定量测定两混合物组分的主要方法有等吸收波长法和系数倍率法两种。
(4)导数分光光度法
采用不同的实验方法可以获得各种导数光谱曲线,包括双波长法、电子微分法和数值微分法。
导数分光光度法对吸收强度随波长的变化非常敏感,灵敏度高。对重叠谱带及平坦谱带的分辨率高,噪声低。可适用于痕量分析以及稀土元素、药物、氨基酸、蛋白质的测定,同时对废气或空气中污染气体的测定也非常有效。
(5)示差分光光度法
用普通分光光度法测定很稀或很浓的溶液的吸光度时,测量误差都很大。若用一已知合适浓度的标准溶液作为参比溶液,调节仪器的100%透光率点(即0吸光度点),测量试样溶液对该已知标准溶液的透光率,则可以改善测量吸光度的精确度,这种方法称为示差分光光度法。
(6)光度滴定法
分光光度滴定法是利用被测组分或滴定剂或反应产物在滴定过程中的吸光度的变化来确定滴定的终点,并由此计算试液中被测组分含量的方法。
(7)其他方法
其他紫外—可见分光光度定量分析方法还包括动力学分光光度法、胶束增溶分光光度法等。
动力学分光光度法是利用反应速率与反应物、产物或催化剂的浓度之间的定量关系,通过测量与反应速率成比例关系的吸光度,从而计算待测物质的浓度。根据催化剂的存在与否,动力学分光光度法可分为非催化和催化分光光度法。
胶束增溶分光光度法是利用表面活性剂的增溶、增敏、增稳、褪色、析相等作用,以提高显色反应的灵敏度、对比度或选择性,改善显色反应条件,并在水相中直接进行光度测量的光度分析法。
4.3.1.4 其他方面的应用
(1)化合物相对分子质量的测定
利用同样生色团骨架的分子λmax及εmax基本相同的特点,若一化合物在紫外—可见光区无吸收,则可将它与另一已知摩尔吸光系数ε的生色团作用形成衍生物。测定一定质量浓度ρ(g·L-1)的该衍生物溶液的吸光度A,可以计算该化合物的相对分子质量,即
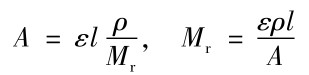
现代岩矿分析实验教程
现代岩矿分析实验教程
式中:Mr为衍生物的相对分子质量,扣除生色团的相对分子质量后得到该化合物的相对分子质量;l为吸收介质厚度(cm)。
(2)氢键强度的测定
溶剂效应对吸收光谱的影响表明,溶剂极性增大,会引起吸收带的蓝移和红移,主要是由于溶质分子与溶剂分子的相互作用而引起的,如果它们之间具有可形成氢键的基团,则是由于形成氢键所引起的,因而可以通过吸收波长的移动程度来测定氢键的强度。
(3)在电化学研究方面的应用
分光光度法与电化学结合,构成了一个崭新的研究领域——光谱电化学。光谱电化学技术包括透射技术、镜反射技术和内反射技术三种。以分光光度法为测量手段,研究某些无机物、有机物和生物物质在电极上的电化学行为,可以同时获得氧化还原体系的吸收光谱和氧化还原电位,以此研究所发生的电化学反应的历程及动力学;还可以测定发生电化学反应所转移的电子数、标准电位、摩尔吸光系数以及反应中间产物或最终产物的扩散系数等。光谱电化学发展很快,在研究无机、有机和生物化学氧化还原机理和均相反应动力学等方面将会发挥极大的作用。
